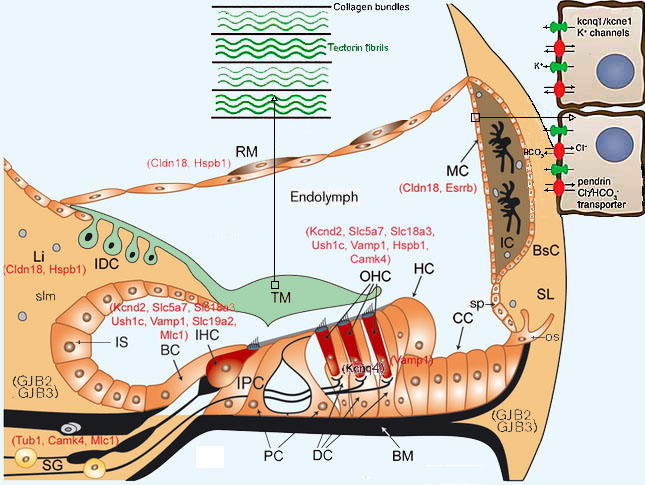
ки. Гены коннексинов (GJB2w GJB3) не экспрессируюгся только в волосковых клетках и клетках спирального возвышения (sp). Калиевые кана-
лы Kcnq4 зкспрессируются только в наружных волосковых клетках (ОНС). Ионы К+, которые поступают во внутренние волосковые клетки (IHC)
возвращаются в эндолимфу по медиальному пути, через клетки внутренней борозды (IS) и фиброциты спирального лимба (slm) к интерденталь-
ным клеткам (IDC). Ионы К+ из наружных волосковых клеток (ОНС) возвращаются в эндолимфу, проходя через клетки Дейтерса (DC), Гензена
(НС), Клаудиса (СС) и клетки наружной борозды (os), затем они проходит через фиброциты спиральной лигаменты (SL) в сосудистую полоску,
состоящую из базальных клеток (BsC), промежуточных клеток (1С) и маргинальных клеток (МС). Остальные обозначения: ВС — пограничные
клетки; ВМ - базилярная мембрана; IDC- интердентальные клетки; IPC - внутренние фалангеапьные клетки; Li - spirallimbus; PC - столбчатые
(pillar) клетки; RM - рейснеровская мембрана; AG — слуховой ганглий; SL— спиральная лигамента; ТМ — текггориальная мембрана
|
условиях между дейтеровскими клетками, внутренними У пациентов с несиндромальной потерей слуха обна- Нейросенсорную глухоту обусловливают также мута- Мутации в гене Сх32 вызывают Х-сцепленную бо- Нарушения плотных соединений Плотные соединения на апикальной поверхности Известно более 20 клоудинов [75]. Локус DFNB29 |
Ген, кодирующий рецептор клоудина 18, estrogen re- Нарушения калиевых каналов Гены Kcnel, Kcnql и Kcnq4 кодируют белковые субъ- Маргинальные клетки сосудистой полоски и вести- Нокаутные по гену Kcnel мыши [24, 110, 132] характе- 10 |
|
membrane translocase), этот короткий белок участвует в Следует также отметить сцепленную с Y-хромосомой Представленные результаты демонстрируют, насколь- 1. Мглинец В.А. Генетические механизмы формированияслуховой улитки и Кортиева органа // Медицинская генетика. - 2011. - Т. 10, №5. - С. 3-14. 2. Abdelhak S., Kalatzis V., Heilig R. et al. A human homologueof the Drosophila eyes absent gene underlies branchio-oto-renal(BOR) syndrome and identifies a novel gene family // Nat. Genet. - 1997. - Vol. 15. - P. 157-164. 3. Adamson C.L., Reid M.A., Mo Z.L. et al. Firing features andpotassium channel content of murine spiral ganglion neurons varywith cochlear location // J. Сотр. Neurol. — 2002. — Vol. 447. —P. 331-350. 4. Ahmad S., Chen S., Sun J., Lin X. Connexins 26 and 30 areсо- assembled to form gap junctions in the cochlea of mice // Biochem. Biophys. Res. Commun. - 2003. - Vol. 307. - P. 362-368. 5. Alsina В., Giraldez F., Pujades C. Patterning and cell fate inear development // Int. J. Dev. Biol. — 2009. — Vol. 53. —P. 1503-1513. 6. Asamura K., Abe S., Imamura Y. et al. Type IX collagen iscrucial for normal hearing // Neuroscience. — 2005. — Vol. 132. —P. 493-500. 7. Ballana E., Ventayol M., Rabionet R. et al. Connexins anddeafness // Homepage. — 2007. — http: // davinci.crg.es/deafness/ 8. Baynash A.G., Hosoda K„ Giaid A. et al. Interaction of endothelin-3 with endothelin-B receptor is essential for developmentof epidermal melanocytes and enteric neurons // Cell. — 1994. —Vol. 79. - P. 1277-1285. 9. Beisel K.W., Nelson N.C., Delimont D.C., Fritzsch B. Longitudinal gradients of KCNQ4 expression in spiral ganglion and cochlear hair cells correlate with progressive hearing loss in DFNA2 //Brain Res. Mol. Brain Res. - 2000. - Vol. 82. - P. 137-149. 10. Beltramello M„ Piazza V,, Bukauskas F.F. et al. Impairedpermeability to Ins(l,4,5)P3 in a mutant connexin underlies recessive hereditary deafness // Nat. Cell Biol. - 2005. — Vol. 7. —P. 63-69. 11. Ben-Yosef Т., Belyantseva I.A., Saunders T.L. et al. Claudin14 knockout mice, a model for autosomal recessive deafnessDFNB29, are deaf due to cochlear hair cell degeneration // Hum.Mol. Genet. - 2003. - Vol. 12. - P. 2049-2061. 12. Bergeron A.L., Schrader A., Yang D. et al. The final stage ofcholinergic differentiation occurs below inner hair cells during development of the rodent cochlea // J. Assoc. Res. Otolaryngol. — 2005. - Vol. 6. - P. 401-415. 13. Berggren D., Frenz D., Galinovic-Schwartz V., Van De Water T.R. Fine structure of extracellular matrix and basal laminae intwo types of abnormal collagen production: L-proline analog-treated
|
14. Bermingham-McDonogh O., Oesterle E.C., Stone J.S. et al.Expression of Proxl During Mouse Cochlear Development //J. Сотр. Neurol. - 2006. - Vol. 496(2). - P. 172-186. 15. Bicego M., Morassutto S., Hernandez V.H. et al. Selectivedefects in channel permeability associated with Cx32 mutations causing X-linked Charcot—Marie—Tooth disease // Neurobiol. Dis. —2006. - Vol. 21. - P. 607-617. 16. Bicego M., Beltramello M., Melchionda S. et al. Pathogenetic role of the deafness-related M34T mutation of Cx26 // Hum.Mol. Genet. - 2006. - Vol. 15. - P. 2569-2587. 17. Blauwkamp M.N., Beyer L.A., Kabara L. et al. The role ofbone morphogenetic protein 4 in inner ear development and function // Hear Res. - 2007. - Vol. 225. - P. 71-79. 18. Boeda В., El-Amraoui A., Bahloul A. et al. Myosin Vila,harmonin and cadherin 23, three Usher 1 gene products that cooperate to shape the sensory hair cell bundle // Embo. J. — 2002. —Vol. 21. - P. 6689-6699. 19. Bok J., Chang W. and Wu D.K. Pattering and morphogenesis of the vertebrate inner ear // Int. J. Dev. Biol. — 2007b. —Vol. 51. - P. 521-533. 20. Bok J., Dolson D.K., Hill P. et al. Opposing gradients of Glirepressor and activators mediate Shh signaling along the dorsoventralaxis of the inner ear // Development. — 2007c. — Vol. 134. —P. 1713-1722. 21. Braunstein E.M., Crenshaw E.B., Morrow B.E., Adams J.C.Cooperative Function of Tbxl and Brn4 in the Periotic Mesenchymeis Necessary for Cochlea Formation // J. Assoc. Res. Otolaryngol. —2008. - Vol. 9. - P. 33-43. 22. Bruzzone R., Cohen-Salmon M. Hearing the messenger:Ins(l,4,5)P3 and deafness // Nat. Cell. Biol. - 2005. - Vol. 7. P. 14-16. 23. Buniello A., Montanaro D., Volinia S. et al. An expressionatlas of connexin genes in the mouse // Genomics. — 2004. —Vol. 83. - P. 812-820. 24. Casimiro M.C., Knollmann B.C., Ebert S.N. et al. Targeteddisruption of the Kcnql gene produces a mouse model of Jervell andLange—Nielsen Syndrome // Proc. Natl. Acad. Sci. USA — 2001. —Vol. 98. - P. 2526-2531. 25. Chang W., ten Dijke P., Wu D.K. BMP pathways are involved in otic capsule formation and epithelial-mesenchymal signalingin the developing chicken inner ear // Dev. Biol. — 2002. —Vol. 251. - P. 380-394. 26. Chen J., Nathans J. Estrogen-related receptor beta/NR3B2controls epithelial cell fate and endolymph production by the striavascularis // Dev. Cell - 2007. - Vol. 13. - P. 325-337. 27. Chen P., Segil N. p27(Kipl) Links Cell Proliferation toMorphogenesis in the Developing Organ of Corti // Development. - 1999. - Vol. 126(8). - P. 1581-1590. 28. Cho H., Yamada Y., Yoo T.J. Ultrastructural changes ofcochlea in mice with hereditary chondrodysplasia (cho/cho) // Ann.N. Y. Acad. Sci. - 1991. - Vol. 630. - P. 259-261. 29. Chu P.J., Rivera J.F., Arnold D.B.A role for Kifl7 in transport of Kv4.2 //J. Biol. Chem. - 2006. - Vol. 281. - P. 365-373. 30. Collin R.W., Kalay E., Tariq M. et al. Mutations of ESRRBencoding estrogen-related receptor beta cause autosomal-recessivenonsyndromic hearing impairment DFNB35 // Am. J. Hum.Genet. - 2008. - Vol. 82. - P. 125-138. 31. Cosgrove D., Meehan D.T., Grunkemeyer J.A. et al. Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome // Genes Dev. - 1996. - Vol. 10. - P. 2981-2992. 32. Cosgrove D., Samuelson G., Meehan D.T. et al. Ultrastructural, physiological and molecular defects in the inner ear of a ge- 14 |
|
70. Kiernan А.Е., Pelling A.L., Leung К.К. et al. Sox2 Is Required for Sensory Organ Development in the Mammalian Inner Ear //Nature. - 2005b. - Vol. 434(7036). - P. 1031-1035. 71. Kiernan A.E., Xu J., Gridley T. The Notch Ligand JAG1 IsRequired for Sensory Progenitor Development in the MammalianInner Ear // PLoS Genet. - 2006. - Vol. 2. - P. e4. 72. Kikuchi Т., Kimura R.S., Paul D.L. et al. Gap junctions inthe rat cochlea: immunohistochemical and ultrastructural analysis //Anat. Embryol. (Berl.). - 1995. - Vol. 191. - P. 101-118. 73. Kirjavainen A., Sulg M., Heyd F. et al. Proxl Interacts withAtohl and Gfil, and Regulates Cellular Differentiation in the InnerEar Sensory Epithelia // Dev. Biol. - 2008. - Vol. 322(1). P. 33-45. 74. Kohlhase J., Wischermann A., Reichenbach H. et al. Mutations in the SALL1 putative transcription factor gene cause Townes—Brocks syndrome // Nat. Genet. — 1998. — Vol. 18. —P. 81-83. 75. Kondoh M., Takahashi A., Fujii M. et al. A novel strategy fora drug delivery system using a claudin modulator // Biol. Pharm.Bull. - 2006. - Vol. 29. - P. 1783-1789. 76. Kubisch C., Schroeder B.C., Friedrich T. et al. KCNQ4, anovel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness // Cell. — 1999. — Vol. 96. —P. 437-446. 77. Kudo Т., Kure S., Ikeda K. et al. Transgenic expression of adominant-negative connexin26 causes degeneration of the organ ofCorti and non-syndromic deafness // Hum. Mol. Genet. — 2003. —Vol. 12. - P. 995-1004. 78. Ladher R.K., Wright T.J., Moon A.M. et al. FGF8 initiatesinner ear induction in chick and mouse // Genes Dev. — 2005. —Vol. 19. - P. 603-613. 79. Lanford P.J., Lan Y., Jiang R. et al. Notch Signalling Pathway Mediates Hair Cell Development in Mammalian Cochlea //Nat. Genet. - 1999. - Vol. 21(3). - P. 289-292. 80. Lanford P.J., Shailam R., Norton C.R. et al. Expression ofMathl and Hes5 in the Cochleae ofWildtype and Jag2 Mutant Mice//J. Assoc. Res. Otolaryngol. - 2000. - Vol. 1(2). - P. 161-171. 81. Le Caignec C., Lefevre M., Schott J.J. et al. Familial Deafness, Congenital Heart Defects, and Posterior Embiyotoxon Causedby Cysteine Substitution in the First Epidermal-Growth-Factor-Like Domain of Jagged 1 //Am. J. Hum.Genet. - 2002. - Vol. 71(1). - P. 180-186. 82. Legan P.K., Lukashkina V.A., Goodyear R.J. et al. A deafness mutation isolates a second role for the tectorial membrane inhearing // Nat. Neurosci. - 2005. - Vol. 8. - P. 1035-1042. 83. Letts V.A., Valenzuela A., Dunbar C. et al. A new spontaneous mouse mutation in the Kcnel gene // Mamm. Genome. —2000. - Vol. 11. - P. 831-835. 84. Li S., Mark S., Radde-Gallwitz K. et al. Hey2 Functions inParallel with Hesl and Hes5 for Mammalian Auditory Sensory Organ Development // BMC Dev. Biol. - 2008. - Vol. 8(1). - P. 20. 85. Li S.W., Takanosu M., Arita M. et al. Targeted disruption ofCollla2 produces a mild cartilage phenotype in transgenic mice:comparison with the human disorder otospondylomegaepiphysealdysplasia (OSMED) // Dev. Dyn. - 2001. - Vol. 222. P. 141-152. 86. Liang J.K., Bok J., Wu D.K. Distinct contributions from thehindbrain and mesenchyme to inner ear morphogenesis // Dev. Biol. - 2010. - Vol. 337. - P. 324-334. 87. Liberman M.C., Tartaglini E., Fleming J.C., Neufeld E.J.Deletion of SLC19A2, the high affinity thiamine transporter, causesselective inner hair cell loss and an auditory neuropathy phenotype// J. Assoc. Res. Otolaryngol. - 2006. - Vol. 7. - P. 211-217. 88. Liu W., Oh S.H., Kang Y.Y. et al. Bone morphogenetic protein 4 (BMP4): a regulator of capsule chondrogenesis in the develo
|
89. Liu X.Z., Ouyang X.M., Xia X.J. et al. Prestin, a cochlearmotor protein, is defective in non-syndromic hearing loss // Hum.Mol. Genet. - 2003. - Vol. 12. - P. 1155-1162. 90. Maddox B.K., Garofalo S., Horton W.A. et al. Craniofacialand otic capsule abnormalities in a transgenic mouse strain with aCol2al mutation // J. Craniofac. Genet. Dev. Biol. — 1998. —Vol. 18. - P. 195-201. 91. Marcus D.C., Sunose H., Liu J. et al. Protein kinase С mediates P2U purinergic receptor inhibition of K+ channel in apicalmembrane of strial marginal cells // Hear. Res. — 1998. — Vol. 115. - P. 82-92. 92. Marcus D.C., Wu Т., Wangemann P., Kofuji P. KCNJ10(Kir4.1) potassium channel knockout abolishes endocochlear potential // Am. J. Physiol. Cell Physiol. - 2002. - Vol. 282. P. C403-407. 93. Maricich S.M., Xia A., Mathes E.L. et al. Atohl-lineal neurons are required for hearing and for the survival of neurons in thespiral ganglion and brainstem accessory auditory nuclei // J. Neurosci. - 2009. - Vol. 29. - P. 11123-11133. 94. McGuirt W.T., Prasad S.D., Griffith A.J. et al. Mutations inCOL11A2 cause non-syndromic hearing loss (DFNA13) // Nat.Genet. - 1999. - Vol. 23. - P. 413-419. 95. Murata J., Tokunaga A., Okano H. et al. Mapping of NotchActivation During Cochlear Development in Mice: Implications forDetermination of Prosensory Domain and Cell Fate Diversification// J. Сотр. Neurol. - 2006. - Vol. 497(3). - P. 502-518. 96. Mustapha M., Weil D., Chardenoux S. et al. An a-tectoringene defect causes a newly identified autosomal recessive form ofsensorineural pre-lingual non-syndromic deafness, DFNB21 //Hum. Mol. Genet. - 1999. - Vol. 8. - P. 409-412. 97. Neyroud N., Tesson F., Denjoy I. et al. A novel mutation inthe potassium channel gene KVLQT1 causes the Jervell and Lange—Nielsen cardioauditory syndrome // Nat. Genet. — 1997. —Vol. 15. - P. 186-189. 98. Nicolas M., Dememes D., Martin A. et al. KCNQ1/KCNE1potassium channels in mammalian vestibular dark cells // Hear. Res. - 2001. - Vol. 153. - P. 132-145. 99. Nowotny M., Gummer A.W. Nanomechanics of the subtectorial space caused by electromechanics of cochlear outer hair cells// Proc. Natl. Acad. Sci. USA. - 2006. - Vol. 103. P. 2120-2125. 100. Ohyama Т., Groves A.K., Martin K. The first steps towardshearing: mechanisms of otic placode induction // Int. J. Dev. Biol. - 2007. - Vol. 51. - P. 463-472. 101. Opdecamp K, Nakayama A., Nguyen M.T. et al. Melanocyte development in vivo and in neural crest cell cultures: crucial dependence on the Mitf basic-helix-loop-helix-zipper transcriptionfactor // Development. - 1997. - Vol. 124. - P. 2377-2386. 102. Pace J.M., Li Y., Seegmiller R.E. et al. Disproportionatemicromelia (Dmm) in mice caused by a mutation in the C-propeptide coding region of Col2al // Dev. Dyn. — 1997. - Vol. 208. P. 25-33. 103. Petersen M.B., Willems P.J. Non-syndromic, autosomal-recessive deafness // Clin. Genet. — 2006. — Vol. 69(5). —P. 371-392. 104. Petersen M.B. Wang Q., Willems P.J. Sex-linked deafness// Clin. Genet. - 2008. - Vol. 73(1). - P. 14-23. 105. Pingault V., Bondurand N., Kuhlbrodt K. et al. SOXIO mutations in patients with Waardenburg—Hirschsprung disease // Nat.Genet. - 1998. - Vol. 18. - P. 171-173. 106. Pirvola U., Ylikoski J., Trokovic R. et al. FGFR1 is required for the development of the auditory sensory epithelium // Neuron. - 2002. - Vol. 35. - P. 671-680. |
|
МЕДИЦИНСКАЯ ГЕНЕТИКА. 2011. №8 107. Puligilla С., Feng F., Ishikawa К. et al. Disruption of fibroblast growth factor receptor 3 signaling results in defects in cellulardifferentiation, neuronal patterning, and hearing impairment // Dev.Dyn. - 2007. - Vol. 236. - P. 1905-1917. 108. Riccomagno M.M., Martinu L., Mulheisen M. et al. Specification of the Mammalian Cochlea Is Dependent on Sonic Hedgehog // Genes Dev. - 2002. - Vol. 16(18). - P. 2365-2378. 109. Richards A.J., Yates J.R., Williams R. et al. A family withStickler syndrome type 2 has a mutation in the COL 11A1 gene resulting in the substitution of glycine 97 by valine in alpha 1 (XI) collagen // Hum. Mol. Genet. - 1996. - Vol. 5. - P. 1339-1343. 110. Rivas A., Francis H.W. Inner ear abnormalities in a Kcnql(Kvlqtl) knockout mouse: a model of Jervell and Lange—Nielsensyndrome // Otol. Neurotol. - 2005. — Vol. 26. — P. 415—424. 111. Rocha-Sanchez S.M., Morris K.A., Kachar B. et al. Developmental expression of Kcnq4 in vestibular neurons and neurosensory epithelia // Brain Res. - 2007. - Vol. 1139. - P. 117-125. 112. Royaux I.E., Bclyantscva I.A., Wu T. et al. Localizationand functional studies of pendrin in the mouse inner ear provide insight about the etiology of deafness in pendred syndrome // J. Assoc.Res. Otolaryngol. - 2003. - Vol. 4. - P. 394-404. 113. Russell I.J., Legan P.K., Lukashkina V.A. et al. Sharpenedcochlear tuning in a mouse with a genetically modified tectorialmembrane // Nat. Neurosci. - 2007. - Vol. 10. - P. 215-223. 114. Sage C., Huang M., Vollrath M. A. et al. Essential role ofretinoblastoma protein in mammalian hair cell development and hearing // PNAS. - 2006. - Vol. 103(19). - P. 7345-7350. 115. Sanchez-Calderon H., Rodriguez-de la Rosa L., Milo M. etal. RNA Microarray Analysis in Prenatal Mouse Cochlea RevealsNovel IGF-I Target Genes: Implication of MEF2 and FOXM1Transcription Factors // PLoS ONE. — 2010. — Vol. 5(1). —P. e8699. 116. Scott D.A., Karniski L.P. Human pendrin expressed in Xenopus laevis oocytes mediates chloride/formate exchange // Am. J.Physiol. Cell Physiol. - 2000. - Vol. 278. - P. 207-211. 117. Shim K., Minowada G., Coling D.E., Martin G.R. Sprouty2, a mouse deafness gene, regulates cell fate decisions in the auditory sensory epithelium by antagonizing FGF signaling // Dev. Cell.- 2005. - Vol. 8. - P. 553-564. 118. Southard-Smith E.M., Kos L., Pavan W.J. Sox 10 mutationdisrupts neural crest development in DomHirschsprung mouse modelI I Nat. Genet. - 1998. - Vol. 18. - P. 60-64. 119. Steel K.P., Barkway C.Another role for melanocytes: theirimportance for normal stria vascularis development in the mammalian inner ear//Development. - 1989. —Vol. 107. - P. 453—463. 120. Suzuki N., Asamura K., Kikuchi Y. et al. Type IX collagenknock out mouse shows progressive hearing loss // Neurosci. Res. —2005. - Vol. 51. - P. 293-298. 121. Tang W., Zhang Y., Chang Q. et al. Connexin29 is highlyexpressed in cochlear Schwann cells and it is required for the normaldevelopment and function of the auditory nerve of mice // J. Neurosci. - 2006. - Vol. 26. - P. 1991-1999. 122. Tassabehji M., Newton V.E., Read A.P. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia(MITF) gene // Nat. Genet. - 1994. - Vol. 8. - P. 251-255. 123. Teijido O., Casaroli-Marano R., Kharkovets T. et al. Expression patterns of MLC1 protein in the central and peripheral nervous systems // Neurobiol. Dis. - 2007. - Vol. 26. - P. 532-545. 124. Teubner В., Michel V., Pesch J. et al. Connexin30 (Gjb6)deficiency causes severe hearing impairment and lack of endocochlear potential // Hum. Mol. Genet. - 2003. - Vol. 12. — P. 13-21. 125. Tiveron M.C., Pattvn A., Hirsch M.R., Brunet J.F. Role ofPhox2b and Mashl in the generation of the vestibular efferent nucleus // Dev. Biol. - 2003. - Vol. 260. - P. 46-57. | 126. Trowe M.O., Maier H., Schweizer M., Kispert A. Deafnessin mice lacking the T-box transcription factor Tbxl8 in otic fibrocytes // Development. - 2008. - Vol. 135. - P. 1725-1734. 127. Tyson J., Tranebjaerg L., Bellman S. et al. IsK andKvLQTl: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell andLange—Nielsen syndrome // Hum. Mol. Genet. — 1997. — Vol. 6. - P. 2179-2185. 128. Usami S., Abe S., Weston M.D. et al. Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused byPDS mutations//Hum. Genet.— 1999,-Vol. 104.-P. 188—192. 129. Vahava O., Morell R., Lynch E.D. et al. Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans // Science. — 1998. — Vol. 279. —P. 1950-1954. 130. Van Camp G., Smith R. Hereditary Hearing Loss Homepage. On World Wide Web URL: http: // dnalab-www.uia.ac.be/dna-lab/hhh/ 131. Verhoeven K.., Van Laer L., Kirschhofer K. et al. Mutations inthe human a-tectorin gene cause autosomal dominant nonsyndromic hearing impairment // Nat. Genet. — 1998. — Vol. 19. — P. 60-62. 132. Vetter D.E., Mann J.R., Wangemann P. et al. Inner ear defects induced by null mutation of the isk gene // Neuron. — 1996. —Vol. 17. - P. 1251-1264. 133. Vikkula M., Mariman E.C., Lui V.C. et al. Autosomal dominant and recessive osteochondrodysplasias associated with theCOL11A2 locus // Cell. - 1995. - Vol. 80. - P. 431-437. 134. Wang J., Mark S., Zhang X. et al. Regulation of polarizedextension and planar cell polarity in the cochlea by the vertebratePCP pathway // Nat. Genet. - 2005. - Vol. 37. - P. 980-985. 135. Wang S.J., Furusho M., D'sa C. et al. Inactivation of fibriblast growth factor receptor signaling in myelinating glial cells result insignificant loss of adult spiral ganglion neurons accompaining byage-related hearing impairment // J. Neurosci. Res. — 2009. —Vol. 87. - P. 3428-3437. 136. Wangemann P., Itza E.M., Albrecht B. et al. Loss ofKCNJ10 protein expression abolishes endocochlear potential andcauses deafness in Pendred syndrome mouse model // BMC Med. —2004. - Vol. 2. - P. 30. 137. Wangemann P., Nakaya K., Wu T. et al. Loss of cochlearHCO3- secretion causes deafness via endolymphatic acidification and inhibition of Ca2+ reabsorption in a Pendred syndrome mouse model //Am. J. Physiol. Renal Physiol. - 2007. - Vol. 292. - P. 1345-1353. 138. Wangemann P. K+ cycling and the endocochlear potential// Hear. Res. - 2002. - Vol. 165. - P. 1-9. 139. Wilcox E.R., Burton Q.L., Naz S. et al. Mutations in thegene encoding tight junction claudin-14 cause autosomal recessivedeafness DFNB29 // Cell. - 2001. - Vol. 104. - P. 165-172. 140. Winter H., Braig C., Zimmermann U. et al. Thyroid hormone receptors TRalphal and TRbeta differentially regulate geneexpression of Kcnq4 and prestin during final differentiation of outerhair cells // J. Cell Sci. - 2006. - Vol. 119. - P. 2975-2984. 141. Woods C., Montcouquiol M., Kelley M.W. Mathl Regulates Development of the Sensory Epithelium in the Mammalian Cochlea // Nat. Neurosci. - 2004. - Vol. 7(12). - P. 1310-1318. 142. Wright T.J., Mansour S.L. Fgf3 and FgflO are required formouse otic placode induction // Development. — 2003. — Vol. 130. - P. 3379-3390. 143. Xiang M., Gan L., Li D. et al. Essential role of POU-domainfactor Bm-Зс in auditory and vestibular hair cell development // Proc.Natl. Acad. Sci. USA. - 1997. - Vol. 94. - P. 9445-9450. 144. Xiang M., Gao W.Q., Hasson Т., Shin J.J. Requirement forBrn-3c in maturation and survival, but not in fate determination ofinnerear hair cells // Development. — 1998. — Vol. 125. —P. 3935-3946. 17 |
|
НАУЧНЫЕ ОБЗОРЫ 145. Yang J.J., Huang S.H., Chou K.H. et al. Identification ofMutations in Members of the Connexin Gene Family as a Cause ofNonsyndromic Deafness in Taiwan // Audiol. Neurootol. — 2007.- Vol. 12. - P. 198-208. 146. Yang S.M., Guo W.W., Hu Y.Y. et al. Smad5 haploinsufficiency leads to hair cell and hearing loss // Dev. Neurobiol. —2009b. - Vol. 69. - P. 153-161. 147. Yang S.M., Hou Z., Yang G. et al. Chondrocyte-SpecificSmad4 Gene Conditional Knockout Results in Hearing Loss and Inner Ear Malformation in Mice // Dev. Dyn. 2009a. — Vol. 238. —P. 1897-1908. 148. Yoshino Т., Sato E., Nakashima T. et al. Distribution of pendrin in the organ of Corti of mice observed by electron immunomicroscopy // Eur. Arch. Otorhinolaryngol. - 2006. - Vol. 263. - P. 699-704. | 149. Zhao H.B., Kikuchi Т., Ngezahayo A., White T.W. Gapjunctions and cochlear homeostasis // J. Membr. Biol. — 2006. —Vol. 209. - P. 177-186. 150. Zheng J.L., Gao W.Q. Overexpression of Mathl InducesRobust Production of Extra Hair Cells in Postnatal Rat Inner Ears //Nat. Neurosci. - 2000. - Vol. 3(6). - P. 580-586. 151. Zine A., Aubert A., Qiu J. et al. Hesl and Hes5 ActivitiesAre Required for the Normal Development of the Hair Cells in theMammalian Inner Ear // J. Neurosci. — 2001. — Vol. 21(13). —P. 4712-4720. 152. Zou D., Erickson C., Kim E.-H. et al. Eyel gene dosage critically affects the development of sensory epithelia in the mammalian inner ear // Human Mol. Genet. - 2008. - Vol. 17(21). - P. 3340-3356. |
1. Genetic infringements different components of cohlea
Mglinets V.A.
Research Centre for Medical Genetics, Russian Academy of Medical Sciences,
115478, 1 Moskvorechie St., Moscow, Russia. E-mail: mglinetz@med-gen.ru
Analyzed genetically caused various forms of syndromal and non-syndromal hearing loss in humans and animal models. Are con-
sidered genetic disorders of individual links in the chain of development and functioning of the organ of hearing. Mutations in these
genes cause both syndromic and non-syndromic deafness, both recessive and dominant forms. Distinguished group of genes that
regulate, for example, extracellular matrix, ion homeostasis and morphogenesis of various types of cells that are important for audi-
tory perception, as well as genes of auditory neuropathy.
Key words: sensorineural deafness, syndromal and non-syndromal hearing loss, genes, mutations
18